医薬品原薬とは原薬を取り巻く法規制
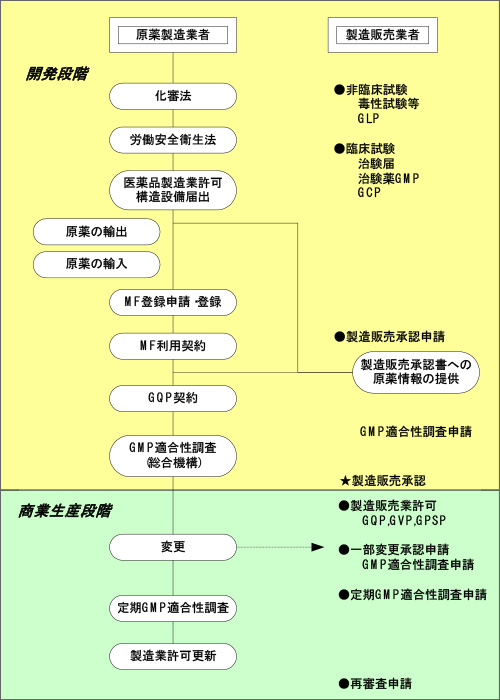
医薬品としての「原薬」の開発から商業生産に係る法規制全体の流れ
医薬品原薬(原薬)は、有機合成、発酵、天然物からの抽出等より得られる化学物質であり、医薬品の有効成分として、品質、有効性、安全性が保証されたものです。
開発段階においては、安全や環境を守るための法律である「化学物質の審査及び製造等の規制に関する法律(化審法)」や「労働安全衛生法」の対象となるとともに、医薬品としては、「医薬品、医療機器等の品質、有効性、安全性の確保等に関する法律(医薬品医療機器等法)」の適用を受けます。
医薬品医療機器等法に従い、原薬製造業者は、製造所毎に厚生労働大臣の許可を受ける必要があります。
原薬は、そのままでは一般消費者に販売されることはなく、製剤化を行うための「製造専用の医薬品」として使用されます。
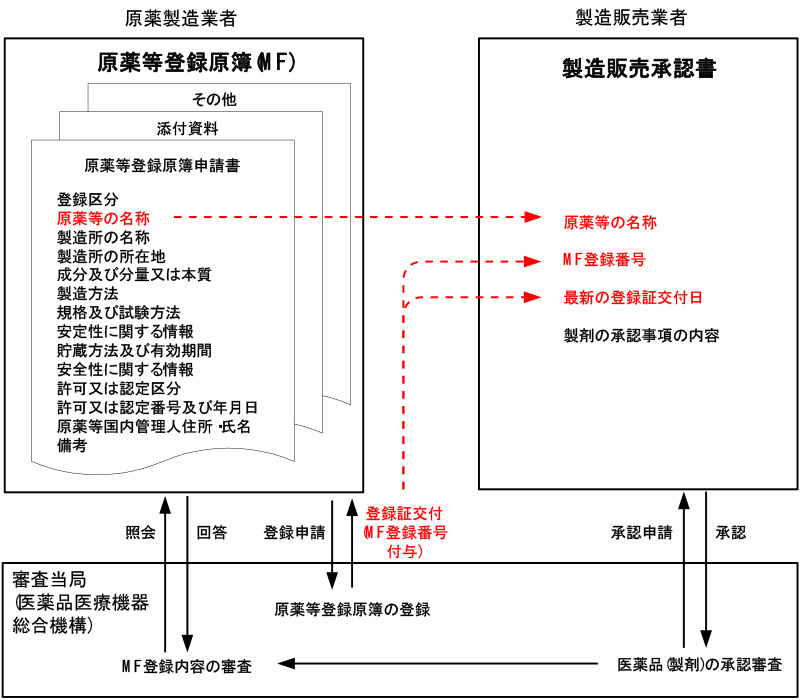
製造販売業者は、製造販売承認申請書に原薬の製造及び品質に関する情報を記載して申請しますが、原薬製造業者が作成し届出た、原薬等登録原簿(マスターファイル:MF)を引用して、製造販売業者が製造販売承認申請を行う場合もあります。
原薬製造業者は、製造販売業者と医薬品の品質管理の基準であるGQPで定められた取決めをもとに、製造販売承認書の記載内容に従って製造管理及び品質管理の基準であるGMPを遵守して製造を行い、行政当局によるGMP適合性調査及び医薬品製造業許可の定期的な更新を受けることになります。
厚生労働大臣の許可とは
国内で原薬を製造するには、薬局等構造設備規則に従い、製造所毎に医薬品の製造業の許可が必要であり、5年毎に更新されることになっています。
マスターファイル(MF)とは
製造販売業者は、原薬の製造及び品質管理に関する情報に関しても製造販売承認申請書に記載しますが、原薬製造業者の知的財産の保護の観点からMF制度が導入されました。
原薬製造業者が原薬の製造方法・製造管理・品質管理等に係る審査に必要な情報を記載したMFを登録し、製造販売業者は、製造販売承認申請書にMF登録番号のみを記載します。
医薬品の製造販売承認申請の審査が開始されるとMFの内容についても審査が行われます。
原薬の製造所におけるGMP適合性調査
医薬品の製造販売承認を受けるには、製造販売承認申請書の審査に加え、原薬を含めた医薬品の製造所がGMP省令を遵守して作業を行っているかについて当局が品目毎に該当する製造所の調査を行い、適合と判断されなければなりません
原薬の輸出入
原薬を輸出入するには、輸出入先国の制度に適合する他、医薬品医療機器等法などの国内の規定に従って手続きを行う必要があります。
原薬の製造に係る医薬品医療機器等法以外の法規制
原薬の製造を行うにあたっては、医薬品を対象とした医薬品医療機器等法の他、以下のような法令及び規則等を遵守しなければなりません。
- 1)化学物質の審査及び製造等の規制に関する法律(化審法)
- 2)特定化学物質の環境への排出量の把握等及び管理の改善の促進に関する法律(化学物質排出把握管理促進法、化管法)
- 3)毒物及び劇物取締法
- 4)労働安全衛生法
- 5)作業環境測定法
- 6)麻薬及び向精神薬取締法
- 7)覚せい剤取締法
- 8)アルコール事業法
- 9)消防法
- 10)高圧ガス保安法
- 11)水質汚濁防止法
- 12)大気汚染防止法
- 13)土壌汚染対策法
- 14)悪臭防止法
- 15)工場立地法
- 16)特定工場における公害防止組織の整備に関する法律
- 17)廃棄物の処理及び清掃に関する法律(廃棄物処理法、廃掃法)
- 18)製造物責任法(PL法)
- 19)遺伝子組換え生物等の使用等の規制による生物の多様性の確保に関する法律(カルタヘナ法)
- 20)外国為替及び外国貿易法(外為法)
- 21)計量法
- 22)特許法